Für die Analyse verglich das Team unter der Leitung von CIBSS-Mitglied Dr. Clemens Kreutz und Prof. Dr. Oliver Schilling fast drei Millionen Auswertungen. Die Forschenden konnten dabei auch zeigen, dass sich bestimmte Open-Source-Programme für die Analyse der komplexen Daten sehr erfolgreich einsetzen lassen. Ihre Ergebnisse sind ein wichtiger Schritt für die künftige Etablierung der Proteinanalyse als diagnostisches Mittel in der klinischen Anwendung und Therapie.
„Unsere Studie zeigt sehr deutlich, dass die Protein-Analyse ein geeignetes Mittel sein kann, um die Diagnostik, beispielsweise zu Krebs, künftig weiter zu verfeinern. Aber auch für viele andere Krankheiten wie Herz-Kreislauf-Erkrankungen könnte diese Form der Diagnostik künftig relevant werden“, sagt Schilling, Heisenberg-Professor für Translationale Proteomics am Institut für Klinische Pathologie des Universitätsklinikums Freiburg.
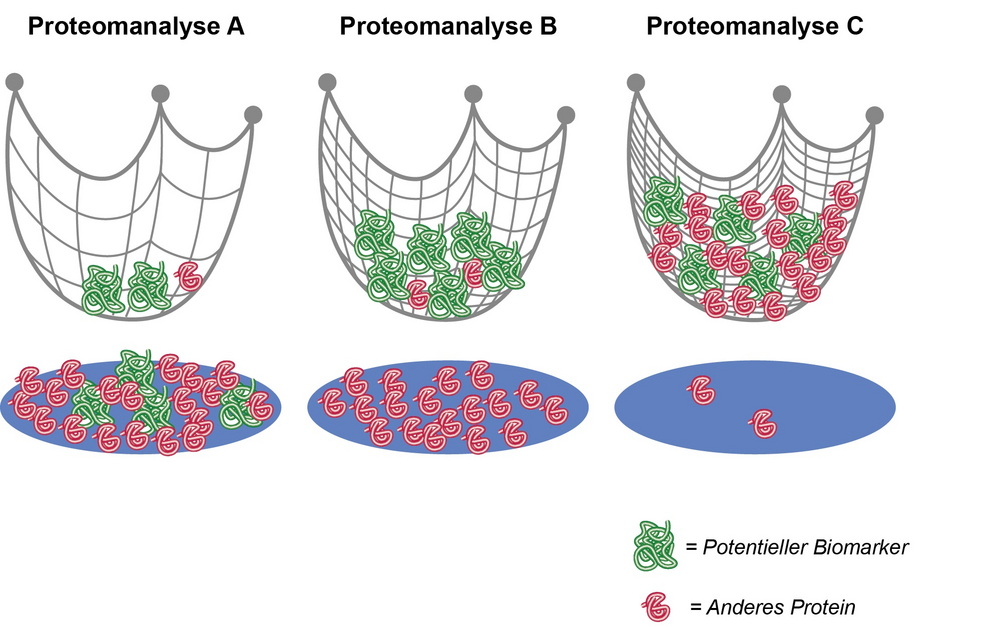
Grundlage für neue Diagnose-Ansätze
Die Forschenden, zu denen auch CIBSS-Mitglied Eva Brombacher gehört, untersuchten Lymphknotengewebe von 92 Krebspatient*innen. Mittels Massenspektrometrie bestimmten sie Typ und Menge von mehr als 7.000 Proteinen. Die Messergebnisse analysierten sie gemeinsam mit Wissenschaftler*innen des Instituts für Biometrie und Statistik des Universitätsklinikums Freiburg und verglichen dabei 1.400 verfügbare Auswertprogramme in jeweils rund 2.100 unterschiedlichen Datensätzen. In der Studie zeigen sie nun, welche der rund 1.400 Kombinationen für die Analyse menschlicher Proben geeignet sind und welche nicht. „Damit haben wir eine wichtige Grundlage für die Entwicklung neuer Diagnostikverfahren geschaffen“, ergänzt Kreutz, Wissenschaftler am CIBSS und Institut für Medizinische Biometrie und Statistik (IMBI) des Universitätsklinikums Freiburg. In internationalen Forschungsverbünden gehen sie weiter der Frage nach, ob sich anhand entsprechend veränderter Proteinmuster der Verlauf oder ein Ansprechen auf eine Therapie absehen lässt.
Originalpublikation:
Fröhlich, K., Brombacher, E., Fahrner, M. et al. Benchmarking of analysis strategies for data-independent acquisition proteomics using a large-scale dataset comprising inter-patient heterogeneity. Nat Commun13, 2622 (2022). https://doi.org/10.1038/s41467-022-30094-0